
Researchers from Johns Hopkins and the University of California, Berkeley have developed a new graph recognition algorithm and toolkit to help address the limitations of the current human reference genome, which researchers use to compare DNA sequences in their studies. Transforming this genomic data into a special graph structure called a “Wheeler graph” provides a more comprehensive and accurate framework for genetic research, the researchers say. The team, which includes Ben Langmead, an associate professor in the Whiting School of Engineering’s Department of Computer Science, published its results in iScience last summer: “WGT: Tools and algorithms for recognizing, visualizing, and generating Wheeler graphs.”
Derived from a small number of similar individuals, the current human reference genome has a Eurocentric bias, which experts say hampers its ability to capture the full genetic diversity of the global population. Using a reference genome that primarily reflects European ethnic groups’ DNA can lead to missing potential birth defects in prenatal screenings, difficulty in diagnosing genetic disorders, and problems in risk assessment for genetic diseases—especially in underrepresented populations.
Ongoing efforts to improve the accuracy and diversity of the human genome face challenges due to its current linear representation, which renders genetic information in only one dimension, like letters in a very long sentence. While this linear genome can be useful for direct DNA comparisons, experts claim that its one-dimensional sequence cannot fully capture the vastness of humanity’s true genetic diversity and complexities.
The Hopkins team says a graph genome, which is more like a series of interconnected webs than a single sentence, ensures a more inclusive and accurate framework for genetic research and medical applications.
“The adoption of a graph genome effectively assimilates data from numerous individuals, providing a more comprehensive representation of intricate genetic variations and accommodating diverse structural changes encountered in various populations,” explains team member Kuan-Hao Chao, a third-year doctoral candidate in Computer Science and a member of the Center for Computational Biology. “This methodology augments precision in genomic analysis, facilitating more accurate sequence mapping and removing reference biases.”
Constructing a graph genome involves transforming diverse genomic data into a structure capable of portraying complex genetic variations and population-specific distinctions, Chao explains. A Wheeler graph, which represents a collection of strings in a way that is particularly easy to index and query, is an ideal structure to use in this scenario.
However, the researchers found that no practical tools to visualize these graphs or to check if existing genomic indexes could be restructured as such were available. To address this gap, they developed a toolkit to generate, visualize, and recognize this kind of graph, with the overarching objective of developing an efficient algorithm for indexing multiple genomes into Wheeler graphs.
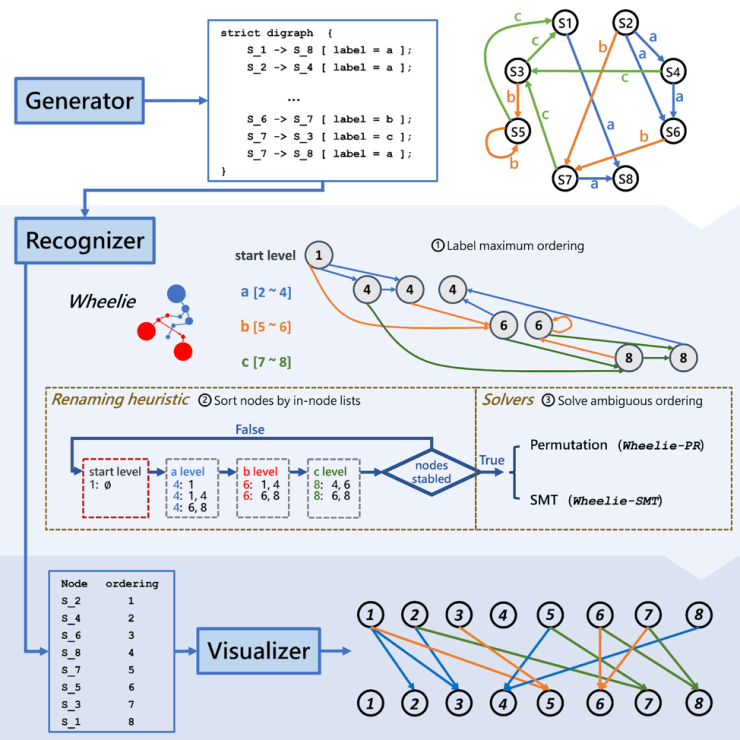
A graphical abstract of the Wheeler Graph Toolkit. First, a user generates a directed graph. The Wheelie algorithm then determines whether or not it is a Wheeler graph before rearranging it into a visual representation of what it would look like in that form.
Their resulting Wheeler Graph Toolkit serves as an open-source suite for creating, identifying, and visualizing Wheeler graphs. At the core of this software lies their “Wheelie” algorithm, which can check whether a given graph has the properties necessary to restructure it into a Wheeler graph. The heuristic algorithm first eliminates impossible combinations to provide an approximate ordering of the graph’s nodes. Wheelie then employs a satisfiability modulo theory solver—which acts like a smart puzzle solver—to determine the final, optimized Wheeler graph ordering.
Through a series of tests and experiments, the team demonstrated that Wheelie is the most practical and robust algorithm available for this graph recognition problem. Effective for graphs with up to 4,000 nodes, 20,000 edges, and five edge labels, Wheelie is also the fastest Wheeler graph recognition algorithm to date, able to check a graph with thousands of nodes within seconds, the researchers say.
Wheelie cannot yet scale to large pangenome graphs, which encompass the entire genetic diversity within a species or population, but the researchers are working on it. They are also tackling an extension of the algorithm that will be capable of correcting any Wheeler graph violations it identifies, thereby transforming a non-Wheeler graph into one for better genomic indexing.
“While we are still far from being able to index multiple genomes into Wheeler graphs, the new ideas introduced in our paper to make the Wheeler graph recognition problem faster and practically computable hold much promise for future research in this area,” remarks Chao.
Additional authors of this work include co-first author Pei-Wei Chen and Sanjit A. Seshia from the University of California, Berkeley.