Spatial transcriptomics (ST)—looking at the spatial arrangement of genes in individual cells—can provide a deeper understanding of how diseases develop and offer clues for potential treatment targets. But one roadblock for this technology is the difficulty of comparing ST data from diverse sources: for example, from tissue samples from healthy versus diseased organs or captured at different scales and resolutions.
Johns Hopkins biomedical engineers have developed a new computational method to precisely align ST data across samples, resolutions, and technologies, enabling researchers to learn more about the underlying biology of cells. According to the team, this new method—called STalign—will improve their ability to make effective comparisons between spatial single-cell data.
“By quantifying the expression of genes in a particular cell, we can better determine the cell’s function and, given their spatial information, understand how these cells are organized in the tissue and work together in shaping health and disease,” said Jean Fan, an assistant professor of biomedical engineering and senior author of the paper. “We hope that this tool can help realize the full potential of spatial transcriptomics for investigating more interesting biological questions.”
The team’s research appears in Nature Communications as “STalign: Alignment of spatial transcriptomics data using diffeomorphic metric mapping.”
The code has been made publicly available via GitHub. The lead authors, Kalen Clifton and Manjari Anant, are doctoral students in Fan’s lab.
Transcriptomic imaging techniques are generally performed on very thin slices of tissue, but the data collection process may squish, tear, and otherwise warp the sample, potentially distorting the spatial positions of cells within the tissue. Likewise, tissue structures may naturally vary from sample to sample or from patient to patient, even if the tissue is taken from the same location in the same organ. This makes analysis all the more challenging, Fan explained, like when trying to compare gene expression and cell composition at similar locations on different tissue samples.
Fan’s team set out to develop a strategy to align tissue slices —even those taken from different ST technologies or patients—to facilitate more accurate spatial molecular comparisons. They collaborated closely with Daniel Tward, an assistant professor of computational medicine and neurology at UCLA, to adapt an algorithm he previously contributed to called large deformation diffeomorphic metric mapping (LDDMM) for aligning MRI images.
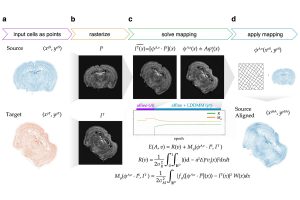
Combining the strengths of machine learning and cutting-edge imaging processing, they developed a novel algorithm to transform cell positions in a source tissue to spatially look more like the cell positions in a target tissue. The researchers tested the algorithm to align mouse brain, human breast cancer, and human heart tissue slices from various ST technologies for comparison at fine resolution.
“We introduce the concept of rasterization to convert the representation of individual cells into a grid of pixels, which drastically improves our ability to perform computations compared to other approaches in the field,” Fan said. “By rasterizing the cells, we don’t have to model the millions of individual cells that are profiled in a spatial transcriptomics experiment. Instead, we can model a smooth image representation that often has a lot fewer pixels. By leveraging LDDMM, we can also perform non-linear alignments that account for local distortions in a tissue sample. This allows us to achieve better structural alignment compared to linear or affine transformations alone.”
The team has made the software publicly available so that other scientists can use the toolkit to align their spatial transcriptomics data. Fan and her team now have their sights set on using STalign to profile tissues from different patients and mouse models, taken at different time points or stages of disease development. The ability to decipher which genes are being expressed at different stages could provide greater insights into conditions ranging from cancer to Alzheimer’s, they say.
“Spatial transcriptomics is poised to be a vital tool in studying diseases,” Fan said. “We hope that having access to STalign will help researchers to apply these technologies and analyze the resulting data effectively and in ways they couldn’t before.”
Additional co-authors include Osagie K. Aimiuwu from the University of North Carolina at Chapel Hill; Michael I. Miller and Justus Kebschull from the Johns Hopkins Department of Biomedical Engineering; and Gohta Aihara and Lyla Atta of the Fan Lab.