



If you’re trying to create a diagnostic test for cancer, you’d expect that the more information you consider, the better. But recent work by Whiting School mathematicians shows that winnowing down the information you’re looking at can be a better strategy.
Advances in gene chip technology now allow researchers to take a tissue sample and easily construct gene expression profiles for thousands of individual genes. Computer programs can then compare the profiles of healthy tissue to cancerous tissue and decide whether a particular kind of cancer is present.
It’s not always easy, and even when the prediction works, it often depends on the combined expression levels of hundreds or even thousands of genes. So the information isn’t much help for researchers looking for clues about what causes the cancer.
Now the winnowing method developed by department chair Daniel Q. Naiman and Donald Geman, both professors in applied mathematics and statistics, is proving itself not only effective but simple enough for humans to understand, apply, and draw hypotheses from.
Rather than consider the levels of hundreds or thousands of genes, their method depends on the relative levels of only two genes. “We decided to go to the other extreme, to do something as simple as possible,” says Geman.
The idea occurred to him when he was reading an article about programmed cell death. It turned out that by finding the levels of only two proteins, called Bax and Bad, you could tell if a cell was programmed to die. If there was more Bax than Bad, the cell would die; otherwise it would live.
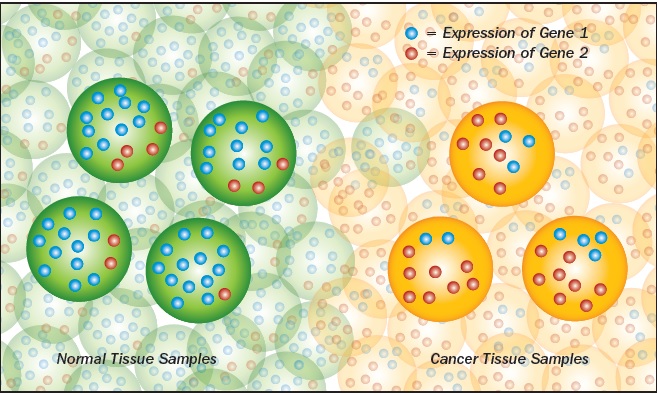
Geman and Naiman wondered if the same thing would work with a test for specific cancers. They obtained microarray data for tissue samples that had been confirmed to be cancerous and took all of the gene-expression levels, threw away the actual numbers, and simply ranked each gene from lowest to highest. Then they compared every gene to every other gene, until they found two genes that by themselves predicted whether a sample was cancerous or not.
“It’s cancer if the expression level of A is less than B. It’s normal if vice versa,” Geman explains.
Although the technique requires a fair amount of number crunching to find the two genes in the first place, it’s something that can be done in a fairly short time on a laptop. And once the pair for each kind of cancer is found, subsequent tests only have to look for those two genes.
Geman and Naiman proposed the idea in a 2004 paper, showing that it worked to predict breast, prostate, and leukemia cancers.
Then earlier this year, researchers from the Institute for Systems Biology in Seattle and the University of Texas in Houston confirmed that the technique also works in telling the difference between two cancers— gastrointestinal stromal tumor and leiomyosarcoma—that can appear similar to diagnosticians but require different treatments. Their results appeared in the Proceedings of the National Academy of Sciences.
And because researchers have an idea of what those two genes do, they have a new lead to follow in trying to understand the underlying mechanism of the cancers.
The technique has its limitations. It doesn’t provide a universal test for cancer—each particular kind of cancer has to be analyzed to see if two genes can be found that predict it. “It’s not a magic bullet,” Geman acknowledges. “So far, none of these methods has made it to the point where it’s part of your blood test.” But stay tuned.